
PHYSIOLOGICAL EFFECTS AND MITOCHONDRIAL DISEASE MODELS
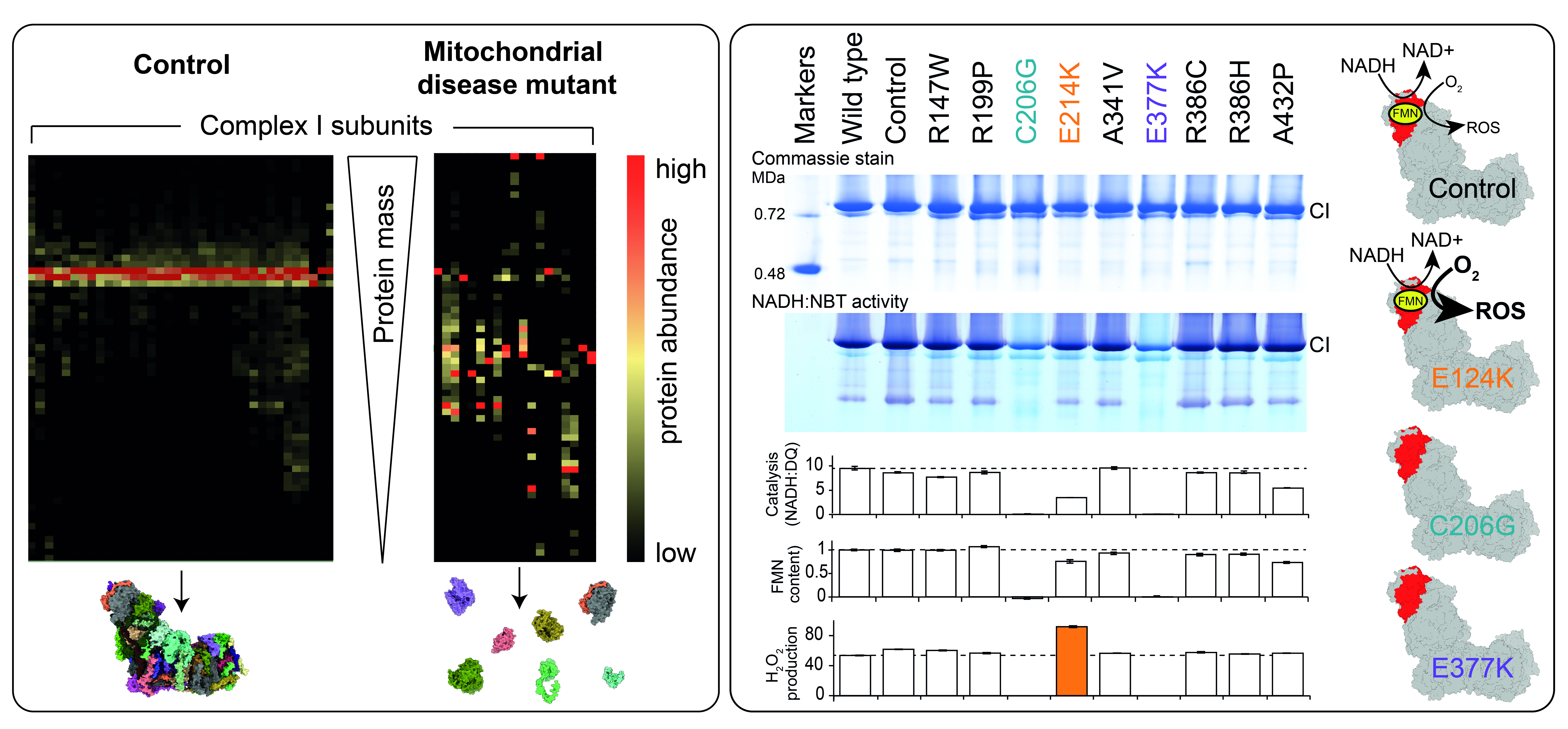
Figure Left; Mass spectrometry ‘complexomics’ analysis of blue-native PAGE from control and mitochondrial disease mutant mitochondrial membranes. Right; Characterisation of human clinical mutations in subunit NDUFV1 replicated in the yeast model Yarrowia lipolytica (adapted from [8]).
Complex I plays a crucial role in the normal function of mitochondrial and cellular metabolism in vivo, and environmental changes and pathogenic mutations can disturb this delicate balance. As well as its catalytic reaction, the enzyme also produces reactive oxygen species from the reduced flavin site [1], [2], contributing to oxidative damage and cellular signalling. Furthermore, under some conditions such as ischaemia, it can also switch between different states and conformations: between the active (ready to catalyse) resting state, and the deactive (requires reactivation) resting state [3]. Complex I-related pathogenic mutations in the nuclear and mitochondrial genome can alter enzyme structure, function, biogenesis and stability, and lead to severe clinical mitochondrial diseases, such as mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), Leber’s hereditary optic neuropathy (LHON) and Leigh syndrome [4]. Their molecular basis may lie in decreased complex I content, for example when protein synthesis or enzyme assembly is impaired [5], or alternatively mutations could yield a normal content of complex I that is compromised in its function. Functional impairment or change can come in the shape of increased or decreased production of reactive oxygen species, reduced rates of catalysis and proton pumping, or a shift in the population of enzyme states in-vivo. An example of the latter behaviour is provided by a mouse mitochondrial mutation model, ND6-P25L, which results in the enzyme rapidly adopting the deactive state in the absence of substrates, an effect that is protective against ischemia reperfusion injury [6]. Our challenge is to work out how genetic mutations cause molecular, mechanistic and structural alterations in complex I, and then how they lead to clinical symptoms, to aid in the understanding, diagnosis and treatment of mitochondrial diseases.
Our research focuses on investigating mitochondrial disease models and the physiological effects of complex I function and dysfunction by:
- Understanding the biochemical and structural properties and roles of the deactive state and the active-deactive transition in complex I, and their relevance in vivo [6], [7];
- Development and application of simple unicellular model systems for studying the molecular effects of mitochondrial disease-causing mutations [8], [9];
- Structural and functional characterisation of complex I in mouse models of mitochondrial disease mutations [6].
References
- Blaza JN, Vinothkumar KR & Hirst J (2018)
Structure of the deactive state of mammalian respiratory complex I.
Structure 26, 312-319 - Kussmaul L & Hirst J (2006)
The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria.
Proc Natl Acad Sci USA 103, 7607-7612 - Pryde KR & Hirst J (2011)
Superoxide is produced by the reduced flavin in mitochondrial complex I: a single, unified mechanism that applies during both forward and reverse electron transfer.
J Biol Chem 286, 18056-18065 - Dröse S, Stepanova A & Galkin A (2016)
Ischemic A/D transition of mitochondrial complex I and its role in ROS generation.
Biochim Biophys Acta 1857, 946-957 - Fassone E & Rahman S. (2012)
Complex I deficiency: clinical features, biochemistry and molecular genetics.
J Med Genet 49, 578-90 - Giachin G, Bouverot R, Acajjaoui S, Pantalone S & Soler-López M (2016)
Dynamics of human mitochondrial complex I assembly: Implications for neurodegenerative diseases.
Front Mol Biosci 3, 43 - Yin Z, Burger N, Kula-Alwar D, Aksentijević D, Bridges HR, Prag HA, Grba DN, Viscomi C, James AM, Mottahedin A, Krieg T, Murphy MP & Hirst J (2021)
Structural basis for a complex I mutation that blocks pathological ROS production.
Nature Commun 12, 707 - Varghese F, Atcheson E, Bridges HR & Hirst J (2015)
Characterization of clinically-identified mutations in NDUFV1, the flavin-binding subunit of respiratory complex I, using a yeast model system.
Hum Mol Genet 24, 6350-6360 - Jarman OD, Biner O, Wright JJ & Hirst J (2021)
Paracoccus denitrificans: a genetically tractable model system for studying respiratory complex I.
Sci Rep 11, 10143