
THE STRUCTURE AND FUNCTION OF ATP SYNTHASES
ROTATION DRIVEN BY ATP HYDROLYSIS
The hydrolytic rotary cycle is composed of three 120o steps in the catalytic F1-domain of the enzyme, defined in biophysical rotation experiments by “catalytic dwells” [1] [2]. Each 120o step is divided into sub-steps defined by a catalytic dwell after each 120o step, with an intervening “ATP binding” dwell at 30o and a “phosphate release” dwell at 95o. Release of phosphate allows the enzyme to complete the 120o step and arrive at the ATP binding dwell. We have described the structures of the F1-catalytic domain at the phosphate release [3] [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] and catalytic [14 ] [15] [16] [17] [18] [19] [20] [21] [22] dwells, and have shown that the 35o rotary sub-step between phosphate release and catalytic dwells depends upon the release of phosphate from the “ATP binding” dwell, which then allows the enzyme to proceed to the “catalytic dwell” [14]. The catalytic dwell itself represents a transition where a bound ATP molecule becomes poised for hydrolysis. Our current efforts are centred around defining the catalytic dwell in the bovine F1-ATPase.
Movie Generation of a 30o rotation of the γ-subunit in bovine F1-ATPase by release of phosphate from the βE-subunit. The movie commences with a view from the side of F1-ThioP in ribbon representation, with the bound thiophosphate as orange spheres. The α- and β-subunits are red and yellow, respectively. Then, the αTP-, αDP-, βTP- and βDP-subunits are removed sequentially leaving the αEβE-dimer and the central stalk subunits γ, δ and ε (blue, magenta and green, respectively). This residual subcomplex is turned to the right around the y-axis by 100° to expose a side-view of the αE-subunit and the central stalk. Then, the δ- and ε-subunits and residues γ-42-210 are removed, and the βE-subunit becomes fainter. In the final frames, the bound phosphate is released and the αE-subunit opens and moves away from the γ-subunit, disrupting packing interactions between the αE- and γ-subunits. In order to re-establish these interactions, the γ-subunit rotates by 30°. The structure of the final state is the post-phosphate release state of bovine F1-ATPase found in F1-I3-ThioP.
The key missing datum for understanding the generation of rotation from the pmf is the detailed structure at atomic level of the pathway the protons take through the membrane domain of the enzyme. The best available structure to date is the one that we determined at 4 Å resolution in 2015 by X-ray crystallography of the enzyme complex from Paracoccus denitrificans [23]. Others were determined in 2015 and 2016 by cryo-electron microscopy of single particles of the bovine and fungal enzymes, at best at 6 Å resolution [24] [25] [26]. However, these models all lack the detail required for formulation of a molecular mechanism of the generation of rotation from the proton-motive force, and, because of the enzymes motility and flexibility, single particles of the enzyme can adopt many different positions. Therefore, it presents an unusually difficult problem for the cryo-em approach.
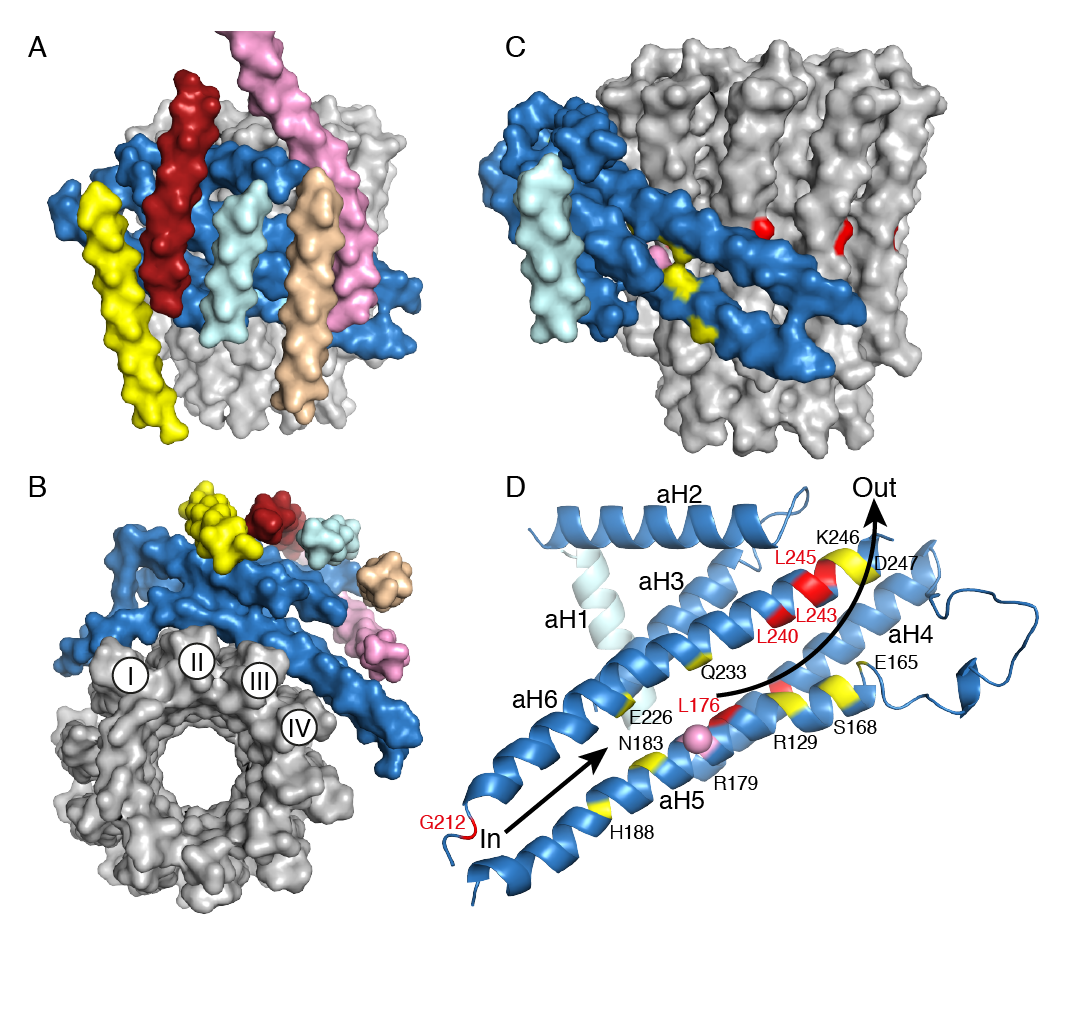
Figure The membrane domain of the F-ATPase from P. angusta. In A-D, the a-subunit is corn-flower blue. A and B, views in solid representation from the side, and below the membrane domain. The c10-ring is grey, the b-subunit (upper part not shown) is pink, and the pale yellow, brick-red, light cyan and beige segments are transmembrane α-helices, Ch1-Ch4 assigned to subunit f, ATP8, aH1 and bH1, respectively. In the c-ring, I-IV indicate the four transmembrane C-terminal α-helices in contact with subunit a. C and D, views of the a-subunit in solid and cartoon representation viewed from outside and looking out from the interface with the c-ring, respectively, with aH1 in pale cyan. Conserved polar residues are yellow, the positions of human mutations associated with pathologies (see Table S1) are red. The pink sphere denotes the conserved Arg-179 in aH5 that is essential for proton translocation. The lower arrow indicates the inlet pathway for protons which transfer to Glu-59 in the C-terminal α-helix-II of the c-ring. They are carried around the ring by anticlockwise rotation as viewed from above, until they arrive at Arg-179 where they enter the exit pathway, denoted by the upper arrow.
STRUCTURES OF BACTERIAL ATP SYNTHASES
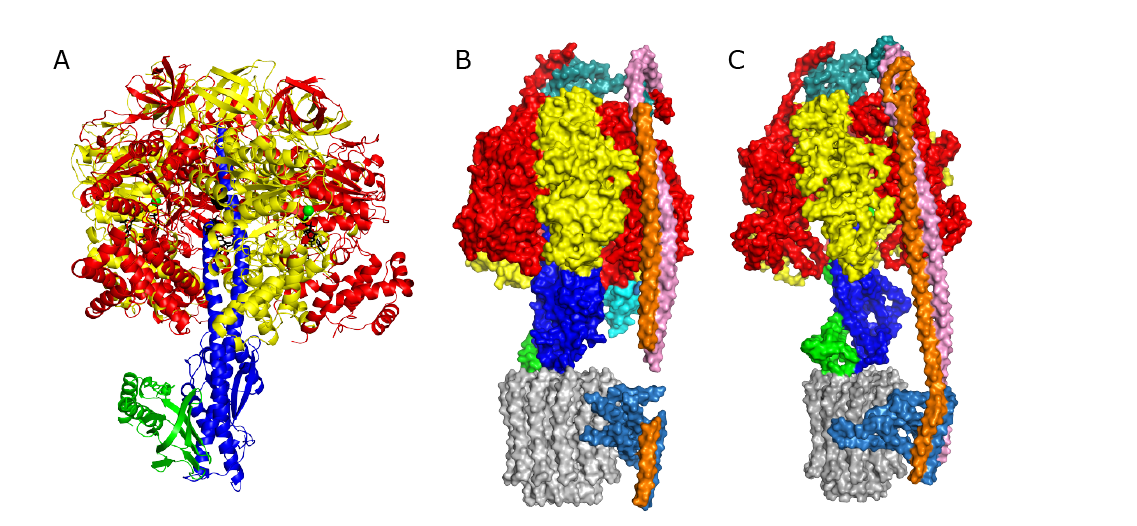
Figure Bacterial ATP synthases. (A) F1-ATPase from Caldalkalibacillus thermarum at 2.6 Å resolution by X-ray crystallography; (B) ATP synthase enzyme from Paracoccus denitrificans at 4 Å resolution by X-ray crystallography; (C) ATP synthase from Escherichia coli at 7 Å resolution by cryo-EM.
Relative to the extensive studies that we have carried out on mitochondrial enzymes, the structures of bacterial ATP synthases have been little investigated, except for structures of the F1 catalytic domains of the enzymes from Escherichia coli [27] and Geobacillus stearothermophilus (formerly Bacillus stearothermophilus) [28] at 3.2 and 3.9 Å resolution, respectively, and structures of the c-rings from their membrane domains from various species [29] [30] [31]. In order to fill this lacuna, so far we have contributed the structure of the entire enzyme from Paracoccus denitrificans at 4 Å resolution [23], and, in collaboration with Professor G. M. Cook and colleagues in Dunedin, New Zealand, the structures of the F1 catalytic domains of the ATP synthases from Caldalkalibacillus thermarum, Fusobacterium nucleatum and Mycobacterium smegmatis. C. thermarum is a thermoalkaliphile, and the structure of its catalytic domain at 2.6 Å resolution is the most accurate structure of a bacterial F1-ATPase yet determined [32]. The enzyme from M. smegmatis is closely related to the enzyme from M. tuberculosis. The structure of its catalytic domain has been determined to 4 Å resolution and provides a target for the development of new anti-tubercular drugs [33]. The opportunistic periodontal pathogen, Fusobacterium nucleatum, is associated with a wide range of diseases including oral infections, advance pregnancy outcomes, gastrointestinal diseases, and atherosclerosis. Also, it has been linked with the development and progression of colorectal cancer via inhibition of anti-tumour immune signalling pathways and subsequent promotion of chemoresistance. It is an obligate anaerobe and couples the free energy of decarboxylation of glutaconyl-CoA to crotonyl-CoA to the transport of Na+ ions across its cellular membrane to generate a sodium ion motive force (smf). The ATP synthase uses the smf to drive the synthesis of ATP required for catabolic and anabolic reactions. The structure of its F1-catalytic domain has been determined at 3.6 Å resolution [34]. Using cryo-EM, a map of the intact ATP synthase of E. coli has been published showing the ε-subunit in the “up” position (see below) [35], and recently, a structure of the ATP synthase from Geobacillus stearothermophilus has been determined to 3 Å [36].
STRUCTURE OF PARASITE ATP SYNTHASES
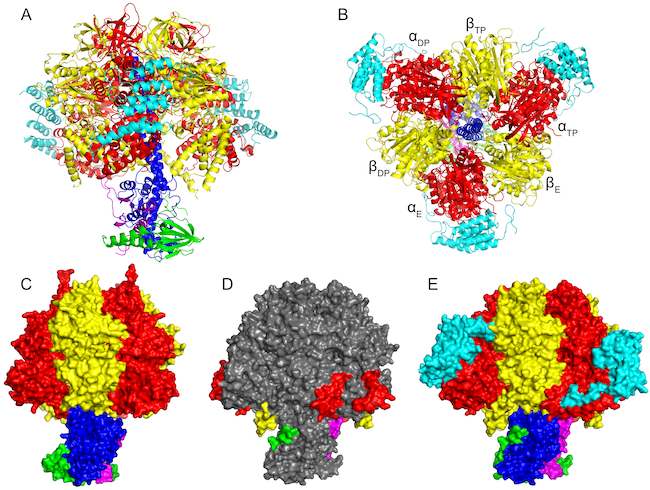
Figure The F1-ATPase from Trypanosoma brucei. The α-, β-, γ-, δ-, ε-, and p18 subunits are red, yellow, blue, green, magenta and cyan respectively. (A and B) Cartoon representations of the side and top views; (C-E) surface representations of (C), the bovine F1-ATPase, (D), the T. brucei enzyme in grey with p18 removed and the coloured sections showing the amino acids additional to the bovine enzyme, and (E), the T. brucei enzyme with p18 present.
The structures and functions of the components of ATP synthases, especially those subunits involved directly in the catalytic formation of ATP, are widely conserved in metazoans, fungi, eubacteria and plant chloroplasts. On the basis of a map at 32.5 Å resolution determined in situ in the mitochondria of the euglenozoan parasite, Trypanosoma brucei, by electron cryo-tomography, it has been proposed that the ATP synthase in this species has a non-canonical structure with different catalytic sites, where the catalytically essential arginine-finger is provided, not by the α-subunit adjacent to the catalytic nucleotide binding site, as in all species investigated to date, but by a protein called p18 found only in the euglenozoa [37]. In a collaboration with Drs Alena Zíková and Ondřej Gahura from the Institute of Parasitology at České Budějovice in the Czech Republic, we have characterized the F1-ATPase from T. brucei [38] and determined a crystal structure of the enzyme at 3.2 Å resolution [39]. This structure shows that the proposal that the catalytic domain of this ATP synthase has a non-canonical structure and different catalytic sites is incorrect. In many respects, the structure of the T. brucei F1-ATPase is closely similar to those of F1-ATPases determined previously. The α3β3-spherical portion of the catalytic domain, where the three catalytic sites are found, plus the central stalk, are highly conserved, and the arginine finger is provided conventionally by the α-subunits adjacent to each of the three catalytic sites found in the β-subunits. Thus, the enzyme has a conventional catalytic mechanism. The structure differs from earlier ones by having a p18-subunit, identified only in the euglenozoa, associated with the external surface of each of the three α-subunits, thereby elaborating the F1-domain. Subunit p18 is a pentatricopeptide repeat (PPR) protein [40] with three PPRs and appears to have no function in the catalytic mechanism of the enzyme. T. brucei is the causative agent of sleeping sickness in humans and related diseases in domestic animals living in Sub-Saharan Africa, and the structure of its F1-ATPases provides a target for development of new drugs for treating the disease.
THE ROLE OF CARDIOLIPIN
The anionic lipid cardiolipin is an essential component of active ATP synthases. In metazoans, their rotors contain a ring of eight c-subunits consisting of inner and outer circles of N- and C-terminal α-helices, respectively. The beginning of the C-terminal α-helix contains a strictly conserved and fully trimethylated lysine residue in the lipid head-group region of the membrane. Larger rings of known structure, from c9-c15 in eubacteria and chloroplasts, conserve either a lysine or an arginine residue in the equivalent position. In computer simulations of hydrated membranes containing trimethylated or unmethylated bovine c8-rings, and bacterial c10- or c11-rings, the head-groups of cardiolipin molecules became associated selectively with these modified and unmodified lysine residues and with adjacent polar amino acids, and with a second conserved lysine on the opposite side of the membrane, whereas phosphatidyl lipids were attracted little to these sites [41].
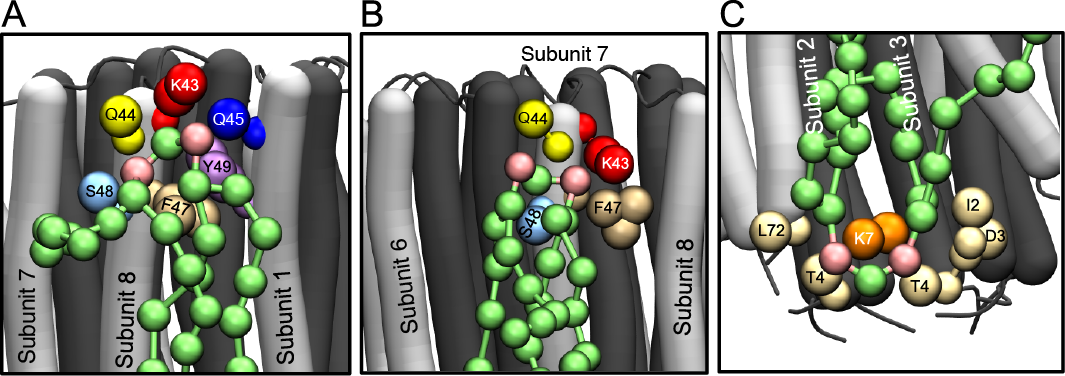
Figure Modes of binding of cardiolipin to trimethylated c8-rings. The N- and C-terminal α-helices of c-subunits are dark and light grey, respectively. Cardiolipin molecules are green, with pink phosphate groups. Large coloured spheres represent the coarse grain beads for specific amino acids, as indicated, lying within 0.7 nm of the phosphate beads of cardiolipin. Parts A and B, the head-group region of cardiolipin in the inner leaflet of the membrane bound, respectively, to two adjacent c-subunits (subunits 8 and 1), and to a single c-subunit; part C, a cardiolipin molecule in the outer leaflet of the membrane bound to a single c-subunit.
However, the residence times of cardiolipin molecules with the ring were brief, and sufficient for the rotor to turn only a fraction of a degree in the active enzyme. With the demethylated c8-ring and with c10- and c11-rings, the density of bound cardiolipin molecules at this site increased, but residence times were not changed greatly. These highly specific but brief interactions with the rotating c-ring are consistent with functional roles for cardiolipin in stabilizing and lubricating the rotor, and, by interacting with the enzyme at the inlet and exit of the transmembrane proton channel, in participation in proton translocation through the membrane domain of the enzyme.
Movie Simulation of interaction of cardiolipin with the native bovine c8-ring. Protein backbone is shown in light grey, with side chains of Lys-43 (red), Gln-44 (yellow), Gln-45 (blue) and Ser-48 (cyan) shown as spheres. Cardiolipin molecules are shown as green sticks, with phosphates as pink spheres; POPC and POPE molecules are shown as dark grey sticks with phosphates as dark grey spheres.
REFERENCES
- Noji H, Yasuda R, Yoshida M & Kinosita K (1997)
Direct observation of the rotation of F1-ATPase.
Nature 386, 299-302 - Yasuda R, Noji H, Yoshida M, Kinosita K & Itoh H (2001)
Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase.
Nature 410, 898-904 - Abrahams JP, Leslie AG, Lutter R & Walker JE (1994)
Structure at 2.8 A resolution of F1-ATPase from bovine heart mitochondria.
Nature 370, 621-628 - van Raaij MJ, Abrahams JP, Leslie AG & Walker JE (1996)
The structure of bovine F1-ATPase complexed with the antibiotic inhibitor aurovertin B.
Proc Natl Acad Sci U S A 93, 6913-6917 - Abrahams JP, Buchanan SK, van Raaij MJ, Fearnley IM, Leslie AG & Walker JE (1996)
The structure of bovine F1-ATPase complexed with the peptide antibiotic efrapeptin.
Proc Natl Acad Sci U S A 93, 9420-9424 - Orriss GL, Leslie AG, Braig K & Walker JE (1998)
Bovine F1-ATPase covalently inhibited with 4-chloro-7-nitrobenzofurazan: the structure provides further support for a rotary catalytic mechanism.
Structure 6, 831-837 - Braig K, Menz RI, Montgomery MG, Leslie AG & Walker JE (2000)
Structure of bovine mitochondrial F(1)-ATPase inhibited by Mg(2+) ADP and aluminium fluoride.
Structure 8, 567-573 - Gibbons C, Montgomery MG, Leslie AG & Walker JE (2000)
The structure of the central stalk in bovine F(1)-ATPase at 2.4 A resolution.
Nat Struct Biol 7, 1055-1061 - Menz RI, Leslie AG & Walker JE (2001)
The structure and nucleotide occupancy of bovine mitochondrial F(1)-ATPase are not influenced by crystallisation at high concentrations of nucleotide.
FEBS Lett 494, 11-14 - Kagawa R, Montgomery MG, Braig K, Leslie AGW & Walker JE (2004)
The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride.
EMBO J 23, 2734-2744 - Bowler MW, Montgomery MG, Leslie AGW & Walker JE (2006)
How azide inhibits ATP hydrolysis by the F-ATPases.
Proc Natl Acad Sci U S A 103, 8646-8649 - Bowler MW, Montgomery MG, Leslie AGW & Walker JE (2007)
Ground state structure of F1-ATPase from bovine heart mitochondria at 1.9 A resolution.
J Biol Chem 282, 14238-14242 - Watt IN, Montgomery MG, Runswick MJ, Leslie AGW & Walker JE (2010)
Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria.
Proc Natl Acad Sci U S A 107, 16823-16827 - Bason JV, Montgomery MG, Leslie AGW & Walker JE (2015)
How release of phosphate from mammalian F1-ATPase generates a rotary substep.
Proc Natl Acad Sci U S A 112, 6009-6014 - Stock D, Leslie AG & Walker JE (1999)
Molecular architecture of the rotary motor in ATP synthase.
Science 286, 1700-1705 - Cabezon E, Montgomery MG, Leslie AGW & Walker JE (2003)
The structure of bovine F1-ATPase in complex with its regulatory protein IF1.
Nat Struct Biol 10, 744-750 - Kabaleeswaran V, Puri N, Walker JE, Leslie AGW & Mueller DM (2006)
Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase.
EMBO J 25, 5433-5442 - Gledhill JR, Montgomery MG, Leslie AGW & Walker JE (2007)
How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria.
Proc Natl Acad Sci U S A 104, 15671-15676 - Kabaleeswaran V, Shen H, Symersky J, Walker JE, Leslie AGW & Mueller DM (2009)
Asymmetric structure of the yeast F1 ATPase in the absence of bound nucleotides.
J Biol Chem 284, 10546-10551 - Rees DM, Montgomery MG, Leslie AGW & Walker JE (2012)
Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F1-ATPase from bovine heart mitochondria.
Proc Natl Acad Sci U S A 109, 11139-11143 - Robinson GC, Bason JV, Montgomery MG, Fearnley IM, Mueller DM, Leslie AGW & Walker JE (2013)
The structure of F₁-ATPase from Saccharomyces cerevisiae inhibited by its regulatory protein IF₁.
Open Biol 3, 120164 - Bason JV, Montgomery MG, Leslie AGW & Walker JE (2014)
Pathway of binding of the intrinsically disordered mitochondrial inhibitor protein to F1-ATPase.
Proc Natl Acad Sci U S A 111, 11305-11310 - Morales-Rios E, Montgomery MG, Leslie AGW & Walker JE (2015)
Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolution.
Proc Natl Acad Sci U S A 112, 13231-13236 - Zhou A, Rohou A, Schep DG, Bason JV, Montgomery MG, Walker JE, Grigorieff N & Rubinstein JL (2015)
Structure and conformational states of the bovine mitochondrial ATP synthase by cryo-EM.
Elife 4, e10180 - Hahn A, Parey K, Bublitz M, Mills DJ, Zickermann V, Vonck J, Kühlbrandt W & Meier T (2016)
Structure of a Complete ATP Synthase Dimer Reveals the Molecular Basis of Inner Mitochondrial Membrane Morphology.
Mol Cell 63, 445-456 - Vinothkumar KR, Montgomery MG, Liu S & Walker JE (2016)
Structure of the mitochondrial ATP synthase from Pichia angusta determined by electron cryo-microscopy.
Proc Natl Acad Sci U S A 113, 12709-12714 - Cingolani G & Duncan TM (2011)
Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation.
Nat Struct Mol Biol 18, 701-707 - Shirakihara Y, Shiratori A, Tanikawa H, Nakasako M, Yoshida M & Suzuki T (2015)
Structure of a thermophilic F1-ATPase inhibited by an ε-subunit: deeper insight into the ε-inhibition mechanism.
FEBS J 282, 2895-2913 - Meier T, Polzer P, Diederichs K, Welte W & Dimroth P (2005)
Structure of the rotor ring of F-Type Na+-ATPase from Ilyobacter tartaricus.
Science 308, 659-662 - Matthies D, Preiss L, Klyszejko AL, Müller DJ, Cook GM, Vonck J & Meier T (2009)
The c13 ring from a thermoalkaliphilic ATP synthase reveals an extended diameter due to a special structural region.
J Mol Biol 388, 611-618 - Preiss L, Langer JD, Yildiz Ö, Eckhardt-Strelau L, Guillemont JEG, Koul A & Meier T (2015)
Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline.
Sci Adv 1, e1500106 - Ferguson SA, Cook GM, Montgomery MG, Leslie AGW & Walker JE (2016)
Regulation of the thermoalkaliphilic F1-ATPase from Caldalkalibacillus thermarum.
Proc Natl Acad Sci U S A 113, 10860-10865 - Zhang AT, Montgomery MG, Leslie AGW, Cook GM & Walker JE (2019)
The structure of the catalytic domain of the ATP synthase from Mycobacterium smegmatis is a target for developing antitubercular drugs.
Proc Natl Acad Sci U S A 116, 4206-4211 - Petri J, Nakatani Y, Montgomery MG, Ferguson SA, Aragão D, Leslie AGW, Heikal A, Walker JE & Cook GM (2019)
Structure of F-ATPase from the obligate anaerobe Fusobacterium nucleatum.
Open Biol 9, 190066 - Sobti M, Smits C, Wong ASw, Ishmukhametov R, Stock D, Sandin S & Stewart AG (2016)
Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states.
Elife 5, e21598 - Guo H, Suzuki T & Rubinstein JL (2019)
Structure of a bacterial ATP synthase.
Elife 8, e43128 - Mühleip AW, Dewar CE, Schnaufer A, Kühlbrandt W & Davies KM (2017)
In situ structure of trypanosomal ATP synthase dimer reveals a unique arrangement of catalytic subunits.
Proc Natl Acad Sci U S A 114, 992-997 - Gahura O, Šubrtová K, Váchová H, Panicucci B, Fearnley IM, Harbour ME, Walker JE & Zíková A (2018)
The F -ATPase from Trypanosoma brucei is elaborated by three copies of an additional p18-subunit.
FEBS J 285, 614-628 - Montgomery MG, Gahura O, Leslie AGW, Zíková A & Walker JE (2018)
ATP synthase fromhas an elaborated canonical F-domain and conventional catalytic sites.
Proc Natl Acad Sci U S A 115, 2102-2107 - Pusnik M, Small I, Read LK, Fabbro T & Schneider A (2007)
Pentatricopeptide repeat proteins in Trypanosoma brucei function in mitochondrial ribosomes.
Mol Cell Biol 27, 6876-6888 - Duncan AL, Robinson AJ & Walker JE (2016)
Cardiolipin binds selectively but transiently to conserved lysine residues in the rotor of metazoan ATP synthases.
Proc Natl Acad Sci U S A 113, 8687-8692