
GENETIC MODIFICATION OF THE MITOCHONDRIAL GENOME
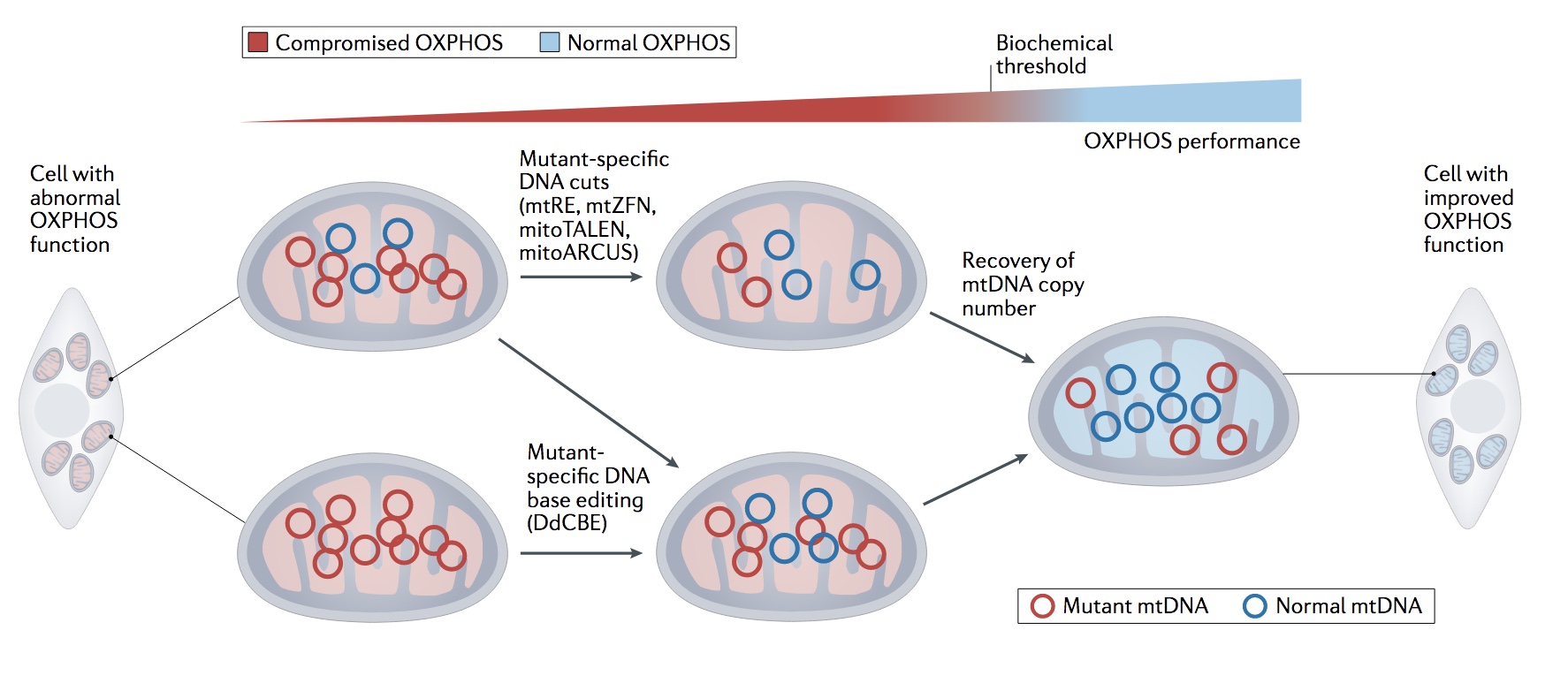
Figure | The general concepts of moving the biochemical threshold in mtDNA disease using mitochondrial genome engineering: selective elimination of the mutant in heteroplasmic populations with nucleases and base editing for homo- and heteroplasmic diseases [1].
Our long-term goal is to develop novel methods for controlled genetic manipulation of mammalian mitochondria in living cells. Several parallel lines of investigation have been pursued in order to identify the most promising approaches [1].
A large part of our work is based upon experiments aimed at developing mitochondrially-targeted engineered nucleases to eliminate human mitochondrial DNA (mtDNA) molecules with pathogenic point mutations and large-scale deletions. One class of designer nuclease technologies utilise the fusion of an engineered zinc finger DNA binding domain to a endonuclease domain, producing a zinc finger-nuclease (ZFN). By targeting pairs of engineered nucleases, adjacently, to a site of interest, a specific DNA double-strand break (DSB) can be produced.
Typically, there are between ~100-10,000 copies of mtDNA per cell. Sequence variation between mtDNA molecules within a cell occurs frequently, a state known as mtDNA heteroplasmy. Often the severity of mitochondrial disease originating from a pathogenic heteroplasmic mutation in mtDNA depends on the ratio of mutated : non-mutated mtDNA present in a cell (Figure, red and green circles, respectively). The heteroplasmic threshold required for disease presentation is variable between mutations, typically requiring ~60-90% mutant mtDNA load. This threshold is dependent on: mutation type, tissue distribution and likely other, as yet unknown factors. A majority of mtDNA-mediated mitochondrial disease is heteroplasmic.
The heteroplasmic nature of mtDNA in the disease state allows an unusual gene therapy approach entailing the selective targeting and degradation of mutant mtDNA molecules, based on DNA sequence discrimination. Reducing mutant mtDNA copy number leads to replication of the spared wild-type molecules, as a result of the cell recovering to the initial, gross mtDNA copy number (Figure) [1].
Our laboratory has developed, and is constantly refining mitochondrially-targeted ZFNs (mtZFNs) to target mammalian mtDNA in situ. After the initial description of an efficient method to deliver ZFPs to mitochondria, we provided proof-of-principle that ZFPs can access and bind sites on human mtDNA with single nucleotide specificity [2][3]. We used mtZFNs to target and cleave mtDNA harbouring the m.8993T>G point mutation associated with neuropathy, ataxia, retinitis pigmentosa (NARP) and the ‘common deletion’ (CD), a 4977bp repeat-flanked deletion associated with Kearns-Sayre and Pearson’s marrow pancreas syndromes [4][5][6]. More recently, we have applied the mtZFN approach in a mammalian in vivo model of mitochondrial disease providing pre-clinical data, useful for future development of treatment for any sufferer of mitochondrial disease owing to heteroplasmic deletion, mutation or rearrangement of mtDNA [7][8][9].
In addition to applying mtZFNs to target and modify heteroplasmy, we are developing and applying novel methods for installation of mtDNA mutations de novo. Currently, we are working on generating animal models of mtDNA rearrangements and using base-editors [10] to introduce point mutations in the mammalian mitochondrial genomes [11].
References
- Silva-Pinheiro P & Minczuk M (2021)
The potential of mitochondrial genome engineering
Nat Rev Genet, doi: 10.1038/s41576-021-00432-x - Minczuk, M, Papworth, MA, Miller, JC, Murphy, MP, Klug, A (2008)
Development of a single-chain, quasi-dimeric zinc-finger nuclease for the selective degradation of mutated human mitochondrial DNA.
Nucleic Acids Res 36, 3926-38 - Minczuk, M, Kolasinska-Zwierz, P, Murphy, MP, Papworth, MA (2010)
Construction and testing of engineered zinc-finger proteins for sequence-specific modification of mtDNA.
Nat Protoc 5, 342-56 - Gammage, PA, Rorbach, J, Vincent, AI, Rebar, EJ, Minczuk, M (2014)
Mitochondrially targeted ZFNs for selective degradation of pathogenic mitochondrial genomes bearing large-scale deletions or point mutations.
EMBO Mol Med 6, 458-66 - Gammage, PA, Gaude, E, Van Haute, L, Rebelo-Guiomar, P, Jackson, CB, Rorbach, J, Pekalski, ML, Robinson, AJ, Charpentier, M, Concordet, J-P, Frezza, C, Minczuk, M (2016)
Near-complete elimination of mutant mtDNA by iterative or dynamic dose-controlled treatment with mtZFNs.
Nucleic Acids Res 44, 7804-16 - Gaude, E, Schmidt, C, Gammage, PA, Dugourd, A, Blacker, T, Chew, SPeak, Saez-Rodriguez, J, O'Neill, JS, Szabadkai, G, Minczuk, M, Frezza, C (2018)
NADH Shuttling Couples Cytosolic Reductive Carboxylation of Glutamine with Glycolysis in Cells with Mitochondrial Dysfunction.
Mol Cell 69, 581-593.e7 - Gammage, PA, Viscomi, C, Simard, M-L, Costa, ASH, Gaude, E, Powell, CA, Van Haute, L, McCann, BJ, Rebelo-Guiomar, P, Cerutti, R, Zhang, L, Rebar, EJ, Zeviani, M, Frezza, C, Stewart, JB, Minczuk, M (2018)
Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo.
Nat Med 24, 1691-1695 - Leslie, M (2018)
'Old' genome editors might treat mitochondrial diseases.
Science 361, 1302 - Brooks, HR (2018)
Mitochondria: finding the power to change.
Cell 175, 891-893 - Mok, BY, de Moraes, MH, Zeng, J, Bosch, DE, Kotrys, AV, Raguram, A, Hsu, F, Radey, MC, Peterson, SB, Mootha, VK, Mougous, JD, Liu, DR (2020).
A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing.
Nature 583, 631-637 - Silva-Pinheiro P, Nash PA, Van Haute L, Mutti CD, Turner K, Minczuk M (2022)
In vivo mitochondrial base editing via adeno-associated viral delivery to mouse post-mitotic tissue.
Nat Commun. 13, 750