
REGULATION OF ATP SYNTHASES
ATP synthases have various mechanisms of inactivation and reactivation of ATP synthesis under conditions of low and increasing available energy. In mitochondria and chloroplasts, when Δp is low, ADP-Mg (without inorganic phosphate) remains bound to one of the three catalytic sites of the enzyme forming an inactive complex. In chloroplasts, it is thought that this inactive ADP inhibited state is stabilized during the hours of darkness by formation of an intramolecular disulfide linkage in the γ-subunit of the ATP synthase. When daylight is restored, the enzyme is reactivated by thioredoxin regulated reduction of the disulfide, and energy dependent dissociation of the ADP-Mg [1] [2].
MITOCHONDRIAL ENZYMES
Mitochondria contain an inhibitor protein known as IF1 [3]. In vitro, at a pH of about 6.7 or below, this protein forms a 1:1 complex with either F1-ATPase or with the intact enzyme complex, the formation of this inhibited complex requiring the hydrolysis of two ATP molecules. In the presence of Δp, the direction of rotation of the rotor reverses, the bound IF1 is released and ATP synthesis resumes. Hence, it has been assumed that the physiological role of IF1 is to prevent the wasteful hydrolysis of ATP in mitochondria under anaerobic conditions.
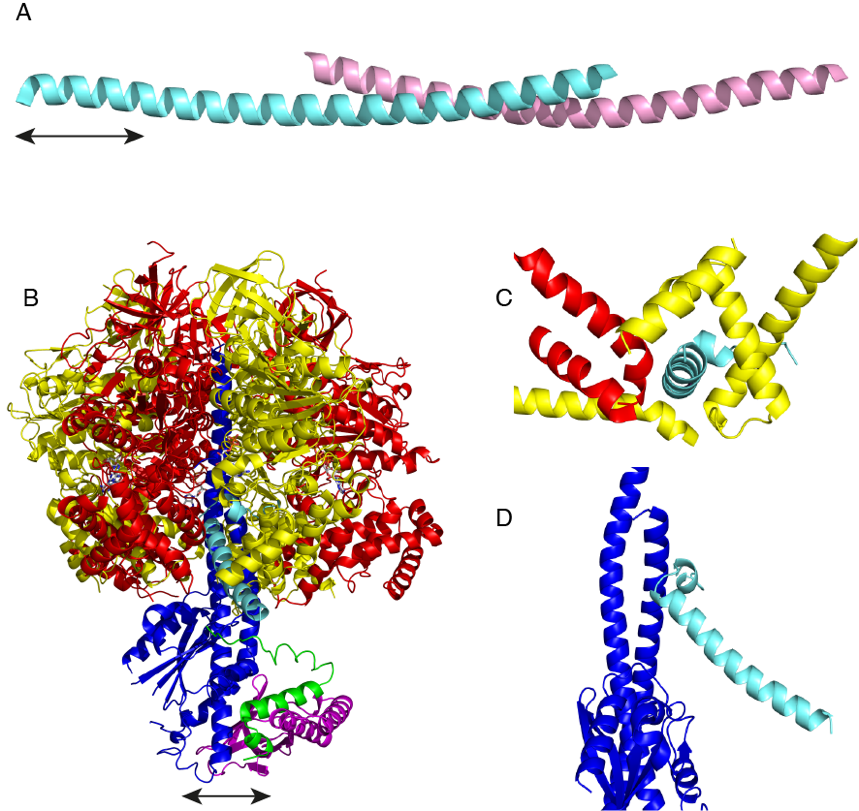
Figure Inhibition of bovine F1-ATPase by IF1.
The mode of inhibition of F1-ATPase by IF1 has been studied in detail. The active bovine protein is a homo-dimer, where the C-terminal regions of the largely α-helical proteins forman antiparallel coiled-coil (Part A) [4]. The protruding N-terminal regions provide the inhibitory part of the protein, and each active dimer can bind to two F1-ATPase moieties [5]. At pH values around 8.0 and higher, the dimers of bovine IF1 form dimers of dimers and higher aggregates, occluding the inhibitory portion of the protein and rendering the inhibitor inactive. Disaggregation of these oligomers into dimers appears to be controlled by the ionic state of a histidine residue, H49, which is thought to provide a pH sensitive switch between inactive and active states [6]. Removal of the capacity to dimerise by deletion of the C-terminal region from residues 61-84 produces a potent monomeric inhibitor that binds to a single F1-ATPase moiety [7]. In some species, for example S. cerevisiae, IF1 lacks the C-terminal dimerization region, and the protein is naturally monomeric [8].
The structures of the monomeric inhibited bovine [7] and yeast F1-IF1 [8] complexes show that IF1 is bound in a complicated binding site at a catalytic interface between the αDP- and βDP-subunits (Panel B). In the bovine complex, the inhibitor protein occupies a deep groove lined with α-helices in the C-terminal domains of the αDP- and βDP-subunits (Panel C), and its N-terminal region interacts with the coiled-coil region of the γ-subunit via a short α-helix (Panel D), and extends into the central aqueous cavity of F1-ATPase. Yeast IF1 binds to yeast F1-ATPase in a similar manner. However, the yeast inhibitor arrests the catalytic cycle of the yeast ATPase at an earlier stage than bovine IF1 inhibits the bovine enzyme. In the yeast inhibited complex, following hydrolysis of ATP the magnesium ion and phosphate have been released, but ADP remains bound to the βE-subunit [8], whereas in the bovine inhibited complex, all three products of hydrolysis have been released from the βE-subunit. The residues in bovine IF1 that provide its binding energy have been identified by systematic mutational analysis and measurement of kinetic constants [9]. Most of the residues are involved in hydrophobic interactions with the C-terminal domain of the βDP-and βTP-subunits. Additional binding energy is provided by an ionic interaction involving glutamate residue E30 of IF1 and arginine residue R408 in the βDP-subunit.
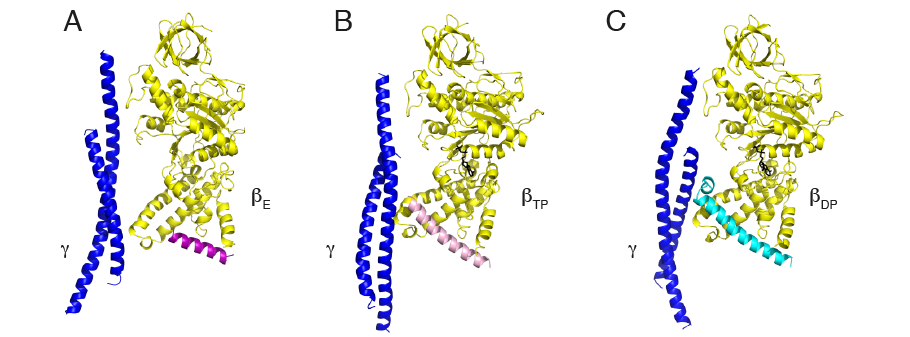
Figure Progressive folding of IF1
One striking feature of the inhibitory region of IF1 is that when the protein is free in solution and not bound to the enzyme, its inhibitory region is intrinsically unfolded and provides an example of an intrinsically disordered protein (or IDP). As the inhibitor binds to the enzyme, this unfolded inhibitory region folds in stages into the α-helical structure observed in the fully inhibited complex. The initial interactions between the unfolded inhibitory region and the ATPase take place in the most open of its three catalytic interfaces, the αEβE-interface, and residues 31-49 of IF1 fold into an α-helix (panel A, above). The hydrolysis of an ATP molecule partially closes this interface and converts it to the αTPβTP-state, allowing more interactions to take place between enzyme and IF1 so that the α-helical region now extends to residues 23-50 (panel B). Hydrolysis of a second ATP molecule converts the αTPβTP-state to the fully closed αDPβDP-state when IF1 forms its most extensively folded state (panel C) described previously [10].
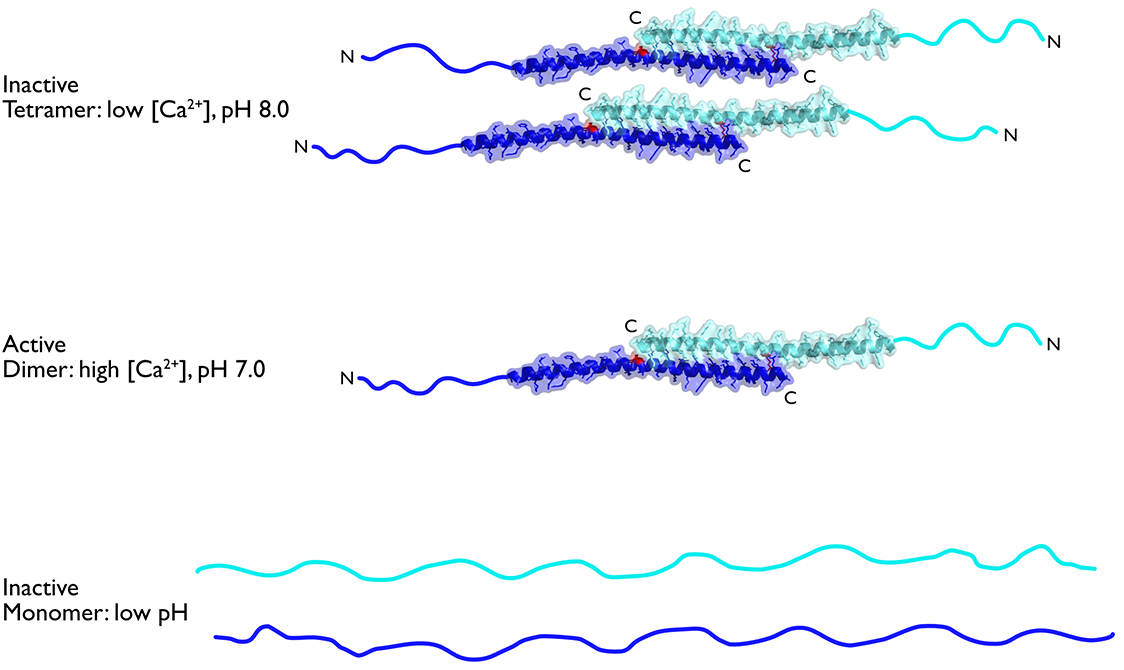
Figure IF1 oligomerisation
It appears from proteomic studies [11] that human mitochondria contain substantial amounts of unbound IF1, even when the ATPase is synthesizing ATP. Therefore, one important question is: how is the inhibitory activity of IF1 itself regulated? IDPs, such as the inhibitory region of IF1, lack a defined structure and populate many different conformational states that are more responsive to solution conditions than folded proteins. Therefore, we investigated the influence of pH and salt types on the self-association of IF1 and the folding of the unfolded region. We found that the equilibrium between dimers and tetramers is probably a central factor in the in vivo modulation of the inhibitory activity. Thus, the intrinsically disordered region makes its inhibitory activity exquisitely sensitive and responsive to physiological changes (in pH and Ca2+ ion concentration, for example) that influence the ability of mitochondria to make ATP [12].
EUBACTERIAL ENZYMES
Eubacteria have evolved a variety of mechanisms for regulating the hydrolytic activity of their ATP synthases. In α-proteobacteria, exemplified by Paracoccus denitrificans, ATP hydrolysis appears to be inhibited by a protein known as the ξ-subunit. The structure of the free ξ-protein in solution has an unfolded N-terminal region followed by bundle of four α-helices in its C-terminal region. Conversely, in the crystal structure of the inhibited ATP synthase complex, the N-terminal region from residues 1-32 is folded into an α-helix, which is bound in catalytic interface of the F1-domain, in a very similar manner to IF1 bound to mitochondrial enzymes, and the C-terminal domain was not detected, presumably because it is mobile [13]. Except for a short region where the ζ-subunit interacts with the bacterial F1-domain, the sequence of the bacterial protein is unrelated to that of IF1.
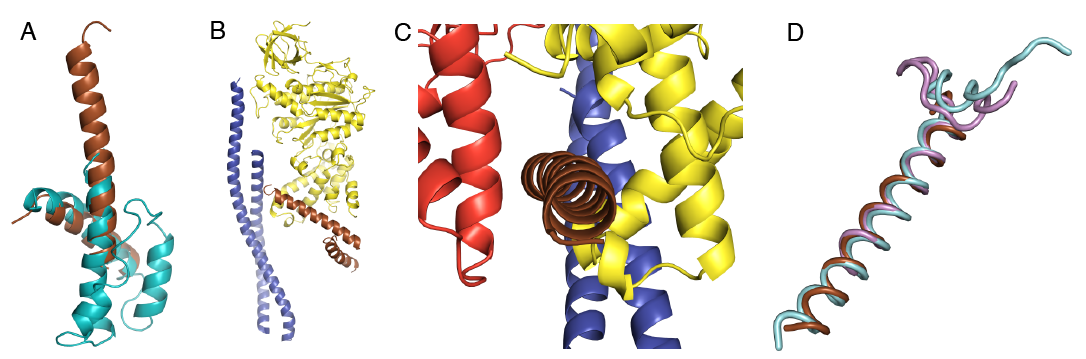
Figure Binding of the ζ-subunit to the ATP synthase from Paracoccus denitrificans. Part A, the ξ-subunit. The brown helices are those resolved in the crystal structure of the ATP synthase from P. denitrificans and the cyan helices are from an NMR structure and are unresolved in the crystal structure. Parts B and C, the ξ-subunit binds in a similar position to IF1. Part D, similarity of the ξ-subunit N-terminal helix (brown) with the bovine and yeast IF1 (light blue and pink, respectively).
The ATP synthases from E. coli, G. stearothermophilus and Thermosynechococcus elongatus can synthesize ATP by oxidative phosphorylation, and, under anoxic conditions, hydrolyze ATP made by glycolysis so as to maintain a pmf required for driving other essential cellular functions, such as transport of nutrients and cell motility. Nonetheless, it has been proposed that in these organisms, when cellular ATP concentration and pmf are low, another inhibitory mechanism may operate to prevent wastage of ATP [14] [15] [16]. It involves the ε-subunits of these enzymes. This subunit, a component of the rotor at the interface between the central stalk and the membrane bound c-ring, is folded into an N-terminal nine-stranded β-sandwich and a C-terminal α-helical hair-pin. The β-sandwich binds the subunit to the γ-subunit and to the c-ring, and the α-helices adopt two conformations “up” and “down”. In the down state, the α-helices bind an ATP molecule, and are associated with the β-sandwich; in the absence of bound ATP the α-helices assume the up position, interact with the α3β3-catalytic domain and inhibit ATP hydrolysis.
Cyanobacterial ATP synthases are regulated by a mechanism that appears to be similar, but not identical, to the way that ATP synthases in the chloroplasts of green plants and algae are regulated. In the absence of light, when the pmf is low, ADP-Mg2+ remains bound to one of the three catalytic sites of the chloroplast enzyme forming an inactive ADP inhibited state of the enzyme [1] [17]. This inhibited state is reinforced by the formation of an intramolecular disulfide bond in the γ-subunit of the enzyme, which is thought to stabilize a β-hairpin structure formed by a unique additional sequence (residues 198-233) in the γ-subunit. This β-hairpin wedges between the β-subunit and the central stalk, and may suppress futile ATP hydrolysis by preventing the rotation of the γ-subunit [18]. When light is restored, the pmf increases, and reduction of the disulfide bond by thioredoxin unlocks the ATP synthetic activity of the enzyme. In cyanobacterial ATP synthases, the γ-subunits also contain a related insertion [19] that appears to inhibit ATP hydrolysis [20], but it lacks the segment containing the two cysteine residues and so it cannot be regulated by a similar redox mechanism [21].
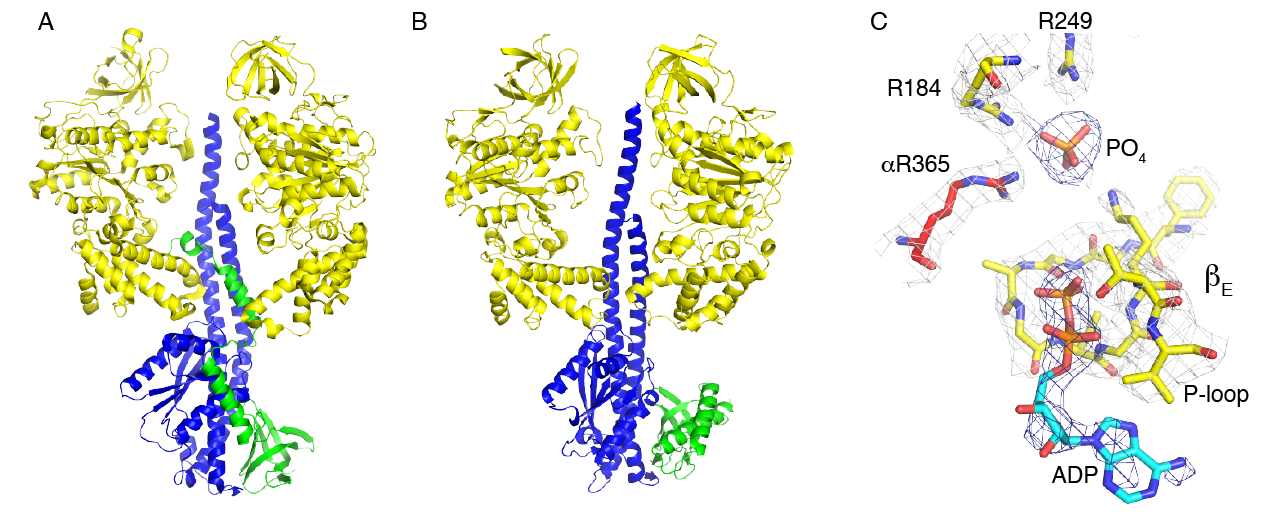
Figure Regulation of F1-ATPase from E. coli and C. thermarum. Parts A and B, comparisson of the positions of the ε-subunit in E. coli and C. thermarum, respectively. The β-subunits, the γ-subunit, and the ε-subunit are cloured yellow, blue and green respectively. The three α-subunits and one of the β-subunits have been removed for clarity. Part C, the nucleotide binding site of βEmpty showing the tightly bound phosphate ion and weakly bound ADP molecule.
The role of the ε-subunit in the regulation of bacterial and chloroplast ATP synthases is an area of active study. The known structures of all bacterial [22] [23] [24] [25] [26] [27] [28] [29], and chloroplast [18][20] ε-subunits, and the orthologous δ-subunit [30] [31] in mitochondria, consist of an N-terminal domain folded into a 10-stranded β-sandwich, and a C-terminal domain folded into an α-helical hair-pin. In the intact enzyme, the β-sandwich domain is involved in binding the ε-subunit to both the γ-subunit in the central stalk of the F1-domain and the c-ring in the membrane domain of the enzyme. In the various structures of ATP synthases and F1-ATPases, the α-helical C-terminal region has been observed in one of two different conformations. In most structures, the two α-helices are associated closely with the β-sandwich, in the “down” conformation, with an ATP molecule bound between the two domains, in the case of C. thermarum (5hkk) [27] and G. stearothermophilus (2e5y) [25]. In the intact ATP synthases from E. coli (5t4o) [28] and G. stearothermophilus (6n2y) [32] and in the F1-domain from E. coli (3ooa) [33] and G. stearothermophilus (2e5y) [26], the two α-helices assume a different “up” conformation, where they penetrate into the α3β3-catalytic domain along the axis of the coiled-coil of the N- and C-terminal α-helices of the γ-subunit [33] [28] [26] [32]. In this conformation, the N-terminal domain has no bound ATP molecule. Therefore, it has been proposed that in the enzyme from G. stearothermophilus [26][32] but not in the E. coli enzyme, that a “down”-“up” switch might provide a physiological mechanism that operates when the pmf and the concentration of ATP are low [34] [35] [36] [25]. Under these conditions, the ATP molecule would leave the ε-subunit, allowing the two α-helices to dissociate from the β-domain and form the inhibitory “up” conformation. In the thermophilic cyanobacterium, T. elongatus, where no nucleotide was observed bound to the isolated subunit (2ro6) [16], and in another structure of the γε subcomplex where no nucleotide was present during the crystallization of the sub-complex (5zwl) [20], the ε-subunit was down in both instances. In this organism it has been proposed that the ATPase is regulated by the γ-subunit, in a similar fashion to the regulation of ATP hydrolysis in the chloroplast enzyme, but without the regulation via the oxidation and reduction of a disulfide linkage that occurs in the chloroplast enzyme [20]. However, the ATP synthases from C. thermarum [27] and M. smegmatis [37] appear not to conform to this mechanism of regulation. They can both synthesize ATP under appropriate conditions, but they hydrolyze ATP very poorly. The structures of their F1-catalytic domains are very similar to each other and also to the F1-domain of the ATP synthase from F. nucleatum, but despite being inhibited in ATP hydrolysis, the ε-subunit of the C. thermarum enzyme is in the “down” position with an ATP molecule and a magnesium ion bound to it. Moreover, the subunit remained in the “down” position and the enzyme remained inhibited when the capacity of the ε-subunit to bind an ATP molecule was removed by mutation [27]. In the mycobacterial enzyme, the α-helical hair-pin of its ε-subunit is truncated and incapable of binding an ATP molecule, and it also is in the “down” position [37]. However, in the structures of the F1-catalytic domains from C. thermarum and M. smegmatis, a phosphate (possibly a sulfate in M. smegmatis) is bound to the most open of the three catalytic sites, suggesting that the hydrolytic activity of this enzyme may be inhibited by the failure to release one of the products of hydrolysis. In the same catalytic site, ADP is bound also in C. thermarum, and is possibly present at low occupancy in M. smegmatis. In F. nucleatum, the ε-subunit is “down” with no bound ATP, but in contrast to the F1-ATPases from C. thermarum and M. smegmatis, it is intrinsically active in ATP hydrolysis, and that activity can be stimulated by LDAO. This behaviour is reminiscent of the behaviour of F1-ATPases from mitochondria, which are similarly intrinsically active and their activity is stimulated also by LDAO. One explanation of the stimulatory effect of LDAO is that it releases inhibitory Mg2+-ADP from the catalytic sites [38] and it has a similar effect on the α3β3γ-subcomplex from G. stearothermophilus [39] [40]. However, in the F1-ATPase from C. thermarum this appears not to be the complete explanation as the activity of this enzyme, in addition to being stimulated by LDAO, is also partially activated by the removal of the C-terminal domain of the ε-subunit and could then be activated to its fullest extent by LDAO [41]. In the E. coli F1-ATPase, where the ε-subunit is permanently “up” [33][42][43], LDAO has an additional effect as it influences interactions between the catalytic β-subunit and the ε-subunit. Thus, currently the most plausible interpretation of the structure of the enzyme from F. nucleatum is that it represents the state of the enzyme that is partially inhibited by Mg2+-ADP, similar to the bovine F1-ATPase, and this partial inhibition can be relieved by LDAO. The exact molecular role of LDAO in activating these various F1-ATPases remains obscure. One possibility is that it loosens the structure of the nucleotide binding domain so that the nucleotide is bound less tightly, and a similar explanation can be advanced to explain the activation of the F. nucleatum F1-ATPase by increased temperatures up to 65oC. Structures of LDAO activated F1-ATPases might help to resolve this issue.
REFERENCES
- Nalin CM & McCarty RE (1984)
Role of a disulfide bond in the gamma subunit in activation of the ATPase of chloroplast coupling factor 1.
J Biol Chem 259, 7275-7280 - Walker JE (1994)
The regulation of catalysis in ATP synthase.
Curr Opin Struct Biol 4, 912-918 - Pullman ME & Monroy GC (1963)
A naturally occurring inhibitor of mitochondrial adenosine triphosphatase.
J Biol Chem 238, 3762-3769 - Cabezón E, Runswick MJ, Leslie AG & Walker JE (2001)
The structure of bovine IF(1), the regulatory subunit of mitochondrial F-ATPase.
EMBO J 20, 6990-6996 - Cabezón E, Arechaga I, Jonathan P, Butler G & Walker JE (2000)
Dimerization of bovine F1-ATPase by binding the inhibitor protein, IF1.
J Biol Chem 275, 28353-28355 - Cabezón E, Butler PJ, Runswick MJ & Walker JE (2000)
Modulation of the oligomerization state of the bovine F1-ATPase inhibitor protein, IF1, by pH.
J Biol Chem 275, 25460-25464 - Gledhill JR, Montgomery MG, Leslie AGW & Walker JE (2007)
How the regulatory protein, IF(1), inhibits F(1)-ATPase from bovine mitochondria.
Proc Natl Acad Sci U S A 104, 15671-15676 - Robinson GC, Bason JV, Montgomery MG, Fearnley IM, Mueller DM, Leslie AGW & Walker JE (2013)
The structure of F₁-ATPase from Saccharomyces cerevisiae inhibited by its regulatory protein IF₁.
Open Biol 3, 120164 - Bason JV, Runswick MJ, Fearnley IM & Walker JE (2011)
Binding of the inhibitor protein IF(1) to bovine F(1)-ATPase.
J Mol Biol 406, 443-453 - Bason JV, Montgomery MG, Leslie AGW & Walker JE (2015)
How release of phosphate from mammalian F1-ATPase generates a rotary substep.
Proc Natl Acad Sci U S A 112, 6009-60014 - Reference not found
- Boreikaite V, Wicky BIM, Watt IN, Clarke J & Walker JE (2019)
Extrinsic conditions influence the self-association and structure of IF, the regulatory protein of mitochondrial ATP synthase.
Proc Natl Acad Sci U S A 116, 10354-10359 - Morales-Rios E, Montgomery MG, Leslie AGW & Walker JE (2015)
Structure of ATP synthase from Paracoccus denitrificans determined by X-ray crystallography at 4.0 Å resolution.
Proc Natl Acad Sci U S A 112, 13231-13236 - Laget PP & Smith JB (1979)
Inhibitory properties of endogenous subunit epsilon in the Escherichia coli F1 ATPase.
Arch Biochem Biophys 197, 83-89 - Kato-Yamada Y, Bald D, Koike M, Motohashi K, Hisabori T & Yoshida M (1999)
Epsilon subunit, an endogenous inhibitor of bacterial F(1)-ATPase, also inhibits F(0)F(1)-ATPase.
J Biol Chem 274, 33991-33994 - Yagi H, Konno H, Murakami-Fuse T, Isu A, Oroguchi T, Akutsu H, Ikeguchi M & Hisabori T (2009)
Structural and functional analysis of the intrinsic inhibitor subunit epsilon of F1-ATPase from photosynthetic organisms.
Biochem J 425, 85-94 - Ketcham SR, Davenport JW, Warncke K & McCarty RE (1984)
Role of the gamma subunit of chloroplast coupling factor 1 in the light-dependent activation of photophosphorylation and ATPase activity by dithiothreitol.
J Biol Chem 259, 7286-7293 - Hahn A, Vonck J, Mills DJ, Meier T & Kühlbrandt W (2018)
Structure, mechanism, and regulation of the chloroplast ATP synthase.
Science 360, eaat4318 - Cozens AL & Walker JE (1987)
The organization and sequence of the genes for ATP synthase subunits in the cyanobacterium Synechococcus 6301. Support for an endosymbiotic origin of chloroplasts.
J Mol Biol 194, 359-83 - Murakami S, Kondo K, Katayama S, Hara S, Sunamura E-I, Yamashita E, Groth G & Hisabori T (2018)
Structure of the γ-ε complex of cyanobacterial F-ATPase reveals a suppression mechanism of the γ subunit on ATP hydrolysis in phototrophs.
Biochem J 475, 2925-2939 - Werner S, Schumann J & Strotmann H (1990)
The primary structure of the gamma-subunit of the ATPase from Synechocystis 6803.
FEBS Lett 261, 204-208 - Wilkens S, Dahlquist FW, McIntosh LP, Donaldson LW & Capaldi RA (1995)
Structural features of the epsilon subunit of the Escherichia coli ATP synthase determined by NMR spectroscopy.
Nat Struct Biol 2, 961-967 - Uhlin U, Cox GB & Guss JM (1997)
Crystal structure of the epsilon subunit of the proton-translocating ATP synthase from Escherichia coli.
Structure 5, 1219-1230 - Wilkens S & Capaldi RA (1998)
Solution structure of the epsilon subunit of the F1-ATPase from Escherichia coli and interactions of this subunit with beta subunits in the complex.
J Biol Chem 273, 26645-26651 - Yagi H, Kajiwara N, Tanaka H, Tsukihara T, Kato-Yamada Y, Yoshida M & Akutsu H (2007)
Structures of the thermophilic F1-ATPase epsilon subunit suggesting ATP-regulated arm motion of its C-terminal domain in F1.
Proc Natl Acad Sci U S A 104, 11233-11238 - Shirakihara Y, Shiratori A, Tanikawa H, Nakasako M, Yoshida M & Suzuki T (2015)
Structure of a thermophilic F1-ATPase inhibited by an ε-subunit: deeper insight into the ε-inhibition mechanism.
FEBS J 282, 2895-2913 - Ferguson SA, Cook GM, Montgomery MG, Leslie AGW & Walker JE (2016)
Regulation of the thermoalkaliphilic F1-ATPase from Caldalkalibacillus thermarum.
Proc Natl Acad Sci U S A 113, 10860-10865 - Sobti M, Smits C, Wong ASw, Ishmukhametov R, Stock D, Sandin S & Stewart AG (2016)
Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states.
Elife 5, e21598 - Joon S, Ragunathan P, Sundararaman L, Nartey W, Kundu S, Manimekalai MSS, Bogdanović N, Dick T & Grüber G (2018)The NMR solution structure of Mycobacterium tuberculosis F-ATP synthase subunit ε provides new insight into energy coupling inside the rotary engine.
FEBS J 285, 1111-1128 - Citekey 869 not found
- Kabaleeswaran V, Puri N, Walker JE, Leslie AGW & Mueller DM (2006)
Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase.
EMBO J 25, 5433-5442 - Guo H, Suzuki T & Rubinstein JL (2019)
Structure of a bacterial ATP synthase.
Elife 8, e43128 - Cingolani G & Duncan TM (2011)
Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation.
Nat Struct Mol Biol 18, 701-707 - Feniouk BA, Suzuki T & Yoshida M (2007)
Regulatory interplay between proton motive force, ADP, phosphate, and subunit epsilon in bacterial ATP synthase.
J Biol Chem 282, 764-772 - Kato S, Yoshida M & Kato-Yamada Y (2007)
Role of the epsilon subunit of thermophilic F1-ATPase as a sensor for ATP.
J Biol Chem 282, 37618-37623 - Suzuki T, Murakami T, Iino R, Suzuki J, Ono S, Shirakihara Y & Yoshida M (2003)
F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of epsilon subunit in response to proton motive force and ADP/ATP balance.
J Biol Chem 278, 46840-46846 - Zhang AT, Montgomery MG, Leslie AGW, Cook GM & Walker JE (2019)
The structure of the catalytic domain of the ATP synthase from Mycobacterium smegmatis is a target for developing antitubercular drugs.
Proc Natl Acad Sci U S A 116, 4206-4211 - Yoshida M & Allison WS (1983)
Modulation by ADP and Mg2+ of the inactivation of the F1-ATPase from the thermophilic bacterium, PS3, with dicyclohexylcarbodiimide.
J Biol Chem 258, 14407-14412 - Matsui T Muneyuki E Honda M Allison W S Dou C & Yoshida M (1997) Catalytic activity of the alpha3beta3gamma of F1-ATPase without noncatalytic nucleotide binding site.
J Biol Chem 272, 8215-8221 - Hirono-Hara Y, Noji H, Nishiura M, Muneyuki E, Hara KY, Yasuda R, Kinosita K & Yoshida M (2001)
Pause and rotation of F(1)-ATPase during catalysis.
Proc Natl Acad Sci U S A 98, 13649-13654 - Keis S, Stocker A, Dimroth P & Cook GM (2006)
Inhibition of ATP hydrolysis by thermoalkaliphilic F1Fo-ATP synthase is controlled by the C terminus of the epsilon subunit.
J Bacteriol 188, 3796-37804 - Sobti M, Smits C, Wong ASw, Ishmukhametov R, Stock D, Sandin S & Stewart AG (2016)
Cryo-EM structures of the autoinhibited E. coli ATP synthase in three rotational states.
Elife 5, e21598 - Sielaff H, Duncan TM & Börsch M (2018)
The regulatory subunit ε in Escherichia coli FF-ATP synthase.
Biochim Biophys Acta Bioenerg 1859, 775-788