
DEFECTS OF MITOCHONDRIAL GENE EXPRESSION IN HUMAN DISEASE
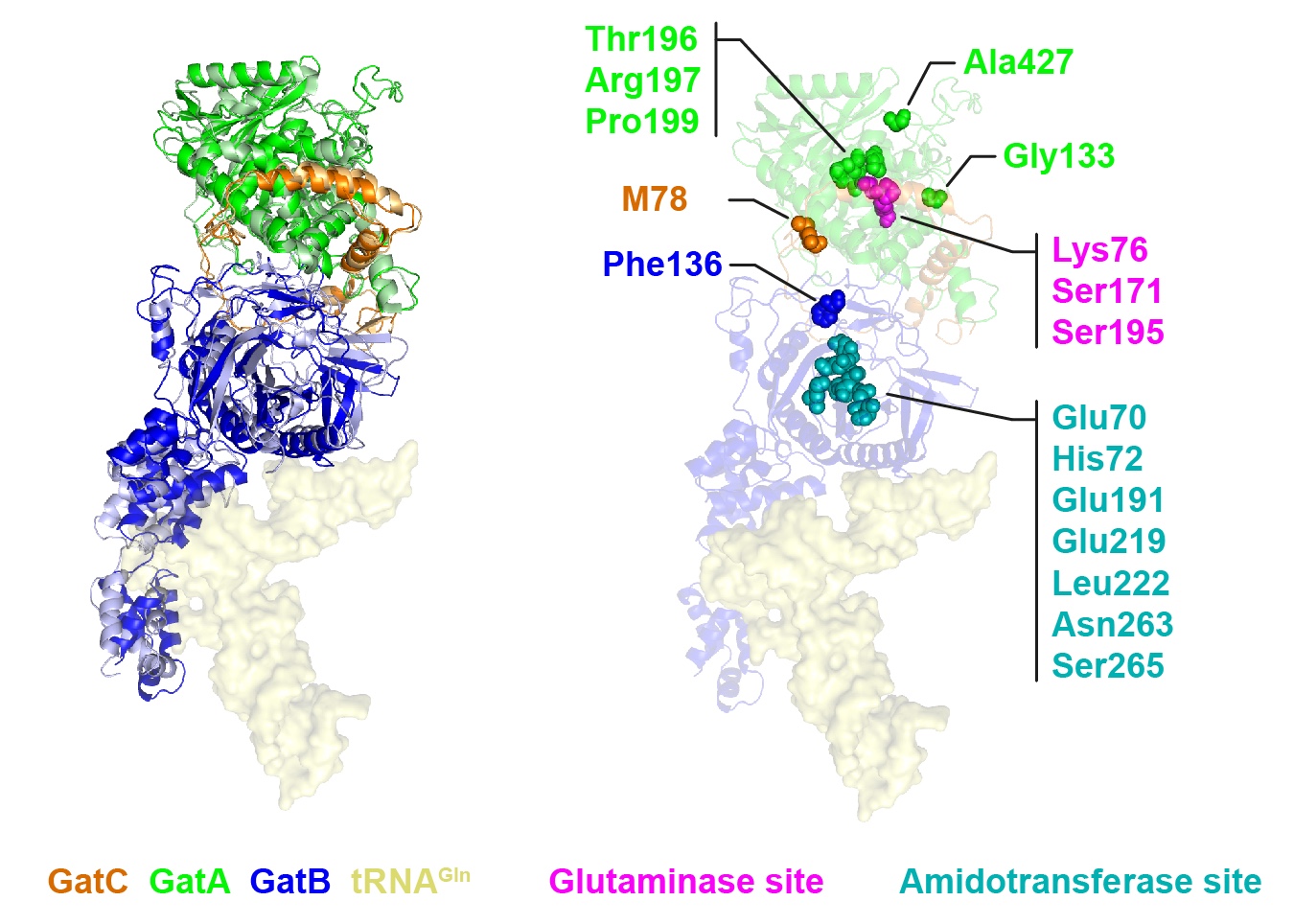
Figure | Three-dimensional model of human mitochondrial GatCAB complex with residues mutated in patients affected with mitochondrial disease.
Much of our understanding of mitochondria has come from studying rare mitochondrial disorders. Thanks to multiple collaborations with clinical groups worldwide, we have been analysing key aspects of mitochondrial genome regulation in samples derived from patients affected with mitochondrial disease. This analysis is a source of valuable insights into the pathomechanisms of human disease, and also into basic mitochondrial molecular genetics. Furthermore, our collaborative studies provide patients with genetic disease with a molecular diagnosis for prevention (prenatal genetic diagnosis) and counseling [1] [2] .
Currently, we are focused on investigating:
Post-transcriptional processing of mitochondrial RNA and translation. We have been studying the ELAC2 gene, coding for an RNase Z responsible for 3’-end processing of mt-tRNAs, in patients with cardiomyopathy, lactic acidosis, and complex I deficiency [3] [4].
Importance of post-transcriptional chamical modification of mitochondrial RNA (epitranscriptomics) for correct expression of mtDNA-encoded genes. We have been analysing several mitochondrial tRNA modifying enzymes, mutations in which often lead to a combined OXPHOS deficiency, cardiomyopathy, lactic acidosis and encephalopathy, including TRMT5 [5] or NSUN3 [6]. More recently we have shown for the first time that defects in the nucleotide modification of mitochondrial ribosomal RNA can lead to human disorders of mitochondrial respiration, by describing variants in the MRM2 gene in a patient with MELAS-like encephalomyopathy [7]. Our studies highlight that defects in mitochondrial translation resulting from incorrect post-transcriptional modification of mt-RNAs are an important contributory factor to the spectrum of human mitochondrial disease [8] [1].
Other aspects of mitochondrial genome maintenance and expression. We identified an orphan gene MGME1 (mitochondrial genome maintenance exonuclease 1, C20orf72) mutated in patients with a mitochondrial syndrome characterised by ocular myopathy, extreme emaciation and respiratory failure associated with mtDNA depletion and large-scale rearrangements. We showed that MGME1 encodes a mitochondrial protein belonging to the PD-(D/E)XK nuclease superfamily, and that MGME1 cleaves single-stranded DNA and processes DNA (and RNA-DNA) flap substrates. Cells from affected individuals show impaired mtDNA replication. This study has demonstrated that MGME1-mediated mtDNA processing is essential for mitochondrial genome maintenance [9] [10] [11]. Our know-how in mt-tRNA metabolism led to a collaboration with the Horvath group on the mechanisms involved in the recovery in reversible infantile respiratory chain deficiency [12] and on a cohort of mitochondrial disease patients harbouring mutations in the mitochondrial transcription elongation factor TEFM (submitted). We also contributed to providing a proof of pathogenicity for more than 20 variants identified in genes involved in aminoacylation of mt-tRNAs (for example: [13]).
REFERENCES
- Van Haute, L, Pearce, SF, Powell, CA, D'Souza, AR, Nicholls, TJ, Minczuk, M (2015)
J Inherit Metab Dis 38, 655-8
-
Nicholls, TJ, Rorbach, J, Minczuk, M (2013)Int J Biochem Cell Biol 45, 845-9
-
Haack, TB, Kopajtich, R, Freisinger, P, Wieland, T, Rorbach, J, Nicholls, TJ, Baruffini, E, Walther, A, Danhauser, K, Zimmermann, FA, Husain, RA, Schum, J, Mundy, H, Ferrero, I, Strom, TM, Meitinger, T, Taylor, RW, Minczuk, M, Mayr, JA, Prokisch, H (2013)Am J Hum Genet 93, 211-23
-
Saoura, M, Powell, CA, Kopajtich, R, Alahmad, A, Al-Balool, HH, Albash, B, Alfadhel, M, Alston, CL, Bertini, E, Bonnen, PE, Bratkovic, D, Carrozzo, R, Donati, MA, Di Nottia, M, Ghezzi, D, Goldstein, A, Haan, E, Horvath, R, Hughes, J, Invernizzi, F, Lamantea, E, Lucas, B, Pinnock, KG, Pujantell, M, Rahman, S, Rebelo-Guiomar, P, Santra, S, Verrigni, D, McFarland, R, Prokisch, H, Taylor, RW, Levinger, L, Minczuk, M. (2019)Hum Mutat 40, 1731-1748.
-
Powell, CA, Kopajtich, R, D'Souza, AR, Rorbach, J, Kremer, LS, Husain, RA, Dallabona, C, Donnini, C, Alston, CL, Griffin, H, Pyle, A, Chinnery, PF, Strom, TM, Meitinger, T, Rodenburg, RJ, Schottmann, G, Schuelke, M, Romain, N, Haller, RG, Ferrero, I, Haack, TB, Taylor, RW, Prokisch, H, Minczuk, MPowell, CA, Kopajtich, R, D'Souza, AR, Rorbach, J, Kremer, LS, Husain, RA, Dallabona, C, Donnini, C, Alston, CL, Griffin, H, Pyle, A, Chinnery, PF, Strom, TM, Meitinger, T, Rodenburg, RJ, Schottmann, G, Schuelke, M, Romain, N, Haller, RG, Ferrero, I, Haack, TB, Taylor, RW, Prokisch, H, Minczuk, M (2015)Am J Hum Genet 97, 319-28
-
Van Haute, L, Dietmann, S, Kremer, L, Hussain, S, Pearce, SF, Powell, CA, Rorbach, J, Lantaff, R, Blanco, S, Sauer, S, Kotzaeridou, U, Hoffmann, GF, Memari, Y, Kolb-Kokocinski, A, Durbin, R, Mayr, JA, Frye, M, Prokisch, H, Minczuk, MA (2016)Nat Commun 7 12039
-
Garone, C, D'Souza, AR, Dallabona, C, Lodi, T, Rebelo-Guiomar, P, Rorbach, J, Donati, MAlice, Procopio, E, Montomoli, M, Guerrini, R, Zeviani, M, Calvo, SE, Mootha, VK, DiMauro, S, Ferrero, I, Minczuk, MA (2017)Hum Mol Genet 26, 4257-4266
-
Powell, CA, Nicholls, TJ, Minczuk, M (2013)Front Genet 6, 79
-
Kornblum, C, Nicholls, TJ, Haack, TB, Schöler, S, Peeva, V, Danhauser, K, Hallmann, K, Zsurka, G, Rorbach, J, Iuso, A, Wieland, T, Sciacco, M, Ronchi, D, Comi, GP, Moggio, M, Quinzii, CM, DiMauro, S, Calvo, SE, Mootha, VK, Klopstock, T, Strom, TM, Meitinger, T, Minczuk, M, Kunz, WS, Prokisch, H (2013)Nat Genet 45, 214-9
-
Nicholls, TJ, Zsurka, G, Peeva, V, Schöler, S, Szczesny, RJ, Cysewski, D, Reyes, A, Kornblum, C, Sciacco, M, Moggio, M, Dziembowski, A, Kunz, WS, Minczuk, M (2014)Hum Mol Genet 23,6147-62
-
Peeva, V, Blei, D, Trombly, G, Corsi, S, Szukszto, MJ, Rebelo-Guiomar, P, Gammage, PA, Kudin, AP, Becker, C, Altmüller, J, Minczuk, MA, Zsurka, G, Kunz, WS (2018)Nat Commun 9, 1727
-
Friederich, MW, Timal, S, Powell, CA, Dallabona, C, Kurolap, A, Palacios-Zambrano, S, Bratkovic, D, Derks, TGJ, Bick, D, Bouman, K, Chatfield, KC, Damouny-Naoum, N, Dishop, MK, Falik-Zaccai, TC, Fares, F, Fedida, A, Ferrero, I, Gallagher, RC, Garesse, R, Gilberti, M, González, C, Gowan, K, Habib, C, Halligan, RK, Kalfon, L, Knight, K, Lefeber, D, Mamblona, L, Mandel, H, Mory, A, Ottoson, J, Paperna, T, Pruijn, GJM, Rebelo-Guiomar, PF, Saada, A, Sainz, B Jr, Salvemini, H, Schoots, MH, Smeitink, JA, Szukszto, MJ, Ter Horst, HJ, van den Brandt, F, van Spronsen, FJ, Veltman, JA, Wartchow, E, Wintjes, LT, Zohar, Y, Fernández-Moreno, MA, Baris,HN, Donnini, C, Minczuk, M, Rodenburg, RJ, Van Hove, JLK (2018)Nat Commun 9:4065.